Sickle cell disease can be deadly, and the persistent health inequities facing Black Americans worsen the problem
Published in News & Features

The pain from a heart attack is so bad that – if you can imagine – it can feel like an elephant sitting on you. Patients with sickle cell disease, a genetic condition affecting the red blood cells, report that this kind of pain begins before their first birthday and continues intermittently for a lifetime.
Too often these people receive inadequate help managing their pain, a critical health disparity that persists 113 years after sickle cell disease was identified.
I am a professor of nursing, and my research focuses on managing pain in sickle cell and cancer patients. I also am a member of the Lancet Haematology Commission, which recently published a report showing paths forward toward worldwide health equity for patients with sickle cell disease.
Here are some key challenges and opportunities I have seen for helping sickle cell patients cope with this disease.
Sickle cell disease results from genetics – more specifically, the inheritance of faulty hemoglobin genes, one from each parent.
Hemoglobin is the iron-rich part of the red blood cell that allows it to carry oxygen from the lungs to all cells in the body.
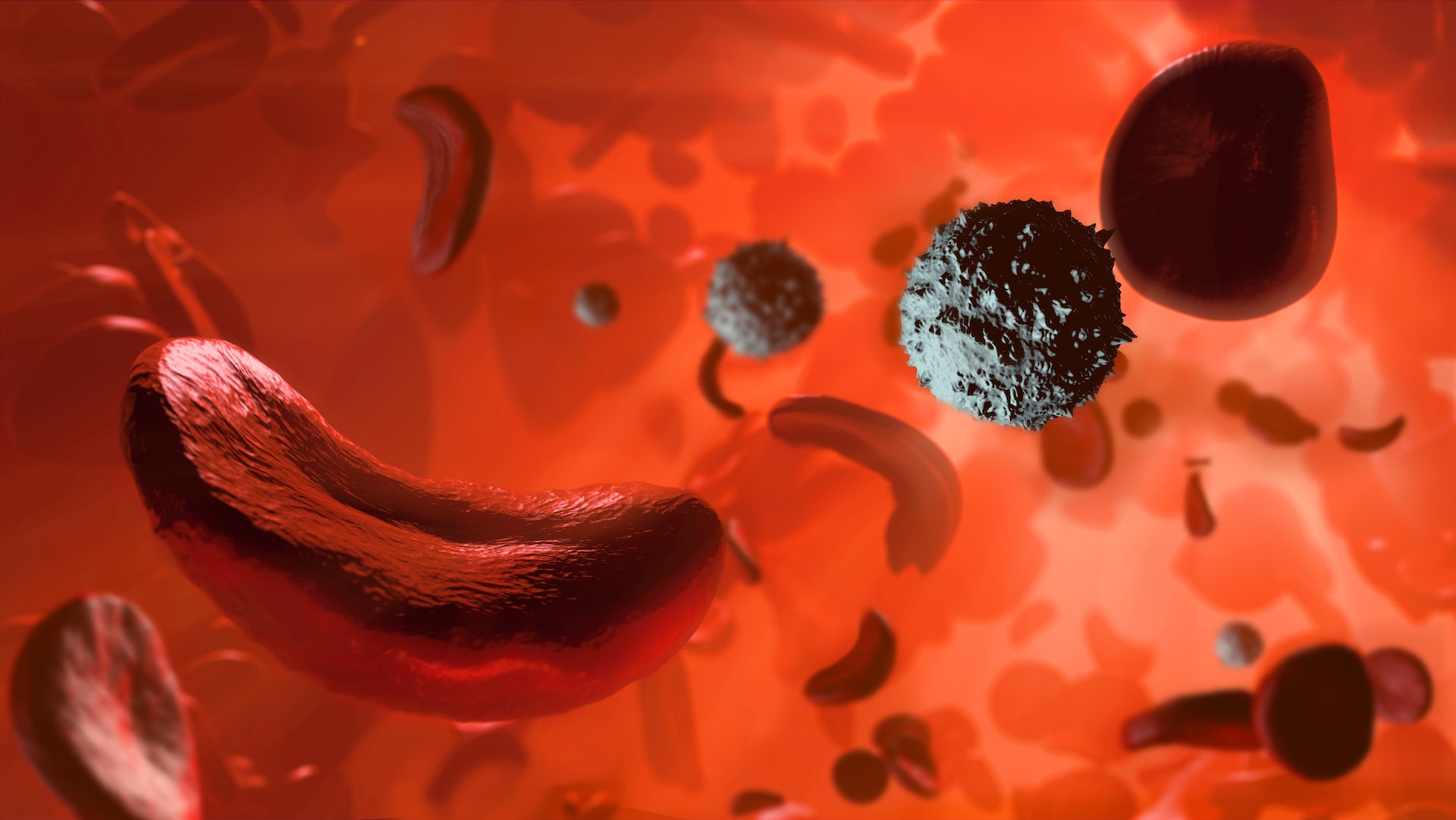
But sickle hemoglobin – known as hemoglobin “S,” which is the faulty hemoglobin gene – causes the typically round red blood cell to be crescent-shaped, like a banana or a sickle. These sickle cells, which are sticky and very stiff, get trapped in tiny blood vessels and block blood flow and oxygen delivery to the body’s organs and tissues.
Not only does this cause pain, it also affects those organs and tissues near the blockages, including the eyes, kidneys, bones, lungs, heart and the brain, where damage can be so severe it causes a stroke. Patients may also have infections, particularly pneumonia and blood infections. Some men with the disease may have painful erections.
For patients with the disease, half of their red blood cells can be sickle cells. Typical red blood cells live for about four months, while sickle cells live for three weeks or less.
This means that patients with sickle cell disease do not have enough cells able to carry oxygen to meet their body’s needs. In the meantime, the slow replacement of cells causes anemia, which is characterized by a reduced number of red blood cells and extreme fatigue.
Worldwide, nearly 8 million people live with sickle cell disease. In the U.S., 100,000 people have the disease. Three million more Americans carry the sickle cell gene, a condition known as sickle cell trait, because they inherited the gene from only one parent.
Most ancestors of those with sickle cell trait likely came from Africa, South Asia or Mediterranean regions where it protected the population against malaria. When the malaria parasite infects the red blood cells of someone with sickle cell trait, the infected cell sickles, and then the body’s immune system removes it.
Sickle cell disease is particularly devastating to families in lower-income countries, where many children with the condition die before their fifth birthday. In contrast, few American children with sickle cell disease who have access to medical care die during childhood.
Still, life expectancy for those with sickle cell disease in the U.S., even with public health insurance, is almost 24 years less than the general population – 52.6 years compared with 76.1 years.
Of the nearly 75,000 Americans hospitalized for sickle cell disease from 2016 to 2018, more than 93% were Black. A century after sickle cell disease was identified, glaring health inequities in its diagnosis, treatment, cures and research about it persist. Although anyone with a faulty hemoglobin gene can pass the disease to their children, in the U.S. it is most common in those of African descent. Structural racism and stigma compound the inequities.
People with the disease frequently visit the emergency room and are hospitalized for pain control. Sometimes, these acute pain episodes are excruciating and last up to 11 days. But often, doctors and nurses do not believe their complaints and requests for strong medications to stop the pain. Instead, they may suspect drug-seeking behavior, deny them pain medicines and label them drug addicts.
These negative views stigmatize those with the disease and contribute to the stress of living with the illness. Patients report feeling “less than human” when providers ignore or discount their pain complaints.
As a result, many patients with sickle cell disease avoid going to the emergency room and do so only when they have exhausted every treatment available at home. They wait, as the pain intensifies to 8, 9 or 10 on a 0-10 scale. By remaining at home, not only is the patient enduring unremitting pain, they are also delaying treatment for organ and tissue damage caused by the disease.
In my view, better dialogue between patients, families and providers is critical. A communications tool called the situation, background, assessment, recommendation communication method could help patients when interacting with providers about their pain and, over time, reduce stigma associated with the illness.
Some adults may have been tested for sickle cell trait as newborns but were not told or do not remember whether they have it. These adults are unaware of their risk for having a baby with the disease. This is why testing, along with genetic counseling, is also critical.
Today in the U.S., all newborn babies are now tested for both sickle cell disease and sickle cell trait. This test helps to spot and treat the disease early, before the baby has symptoms or serious problems, and reduces one structural racism contributor because every baby is tested.
Red blood cell transfusions are an important and lifesaving treatment for sickle cell disease. But there are challenges to transfusion therapy, including difficulty getting into a vein, iron overload, reactions to the transfusion and the time commitment and travel expenses.
Treatment with daily oral hydroxyurea is the standard of care for prevention of pain in both children and adults with sickle cell. This drug increases fetal hemoglobin, which reduces red blood cell sickling. There are also other drugs available that can reduce red blood cell sickling.
One therapy that can cure sickle cell disease is hematopoietic stem cell transplantation. This is the transfer of stem cells capable of making normal blood cells to replace the sickle cells. The stem cells are transplanted to the patient from a well-matched sibling, which limits who can get them.
Since 1984, 1,200 hematopoietic stem cell transplantation cases have been reported. However, the treatment can have serious toxicities, fatal complications, effects on fertility and significant financial costs that must be weighed against the severity of the disease.
Other cures are gene therapies to repair the faulty hemoglobin gene so that it no longer makes sickle red blood cells. But gene therapy is not yet a standard cure.
Neither of these curative treatments alters the genes in the ovum or sperm, so the patient’s future children are still at risk of inheriting the disease.
A future with health equity for sickle cell disease is possible, but only when pain is controlled and treatments have fewer complications and can be made affordable everywhere.
This article is republished from The Conversation, an independent nonprofit news site dedicated to sharing ideas from academic experts. If you found it interesting, you could subscribe to our weekly newsletter.
Read more:
Anemia afflicts nearly 1 in 4 people worldwide, but there are practical strategies for reducing it
Including race in clinical algorithms can both reduce and increase health inequities – it depends on what doctors use them for
Diana J. Wilkie receives funding from the National Institutes of Health. She collaborates with many scientists and clinicians focused on sickle cell disease. She was a member of the Lancet Haematology Commission on Sickle Cell Disease.







Comments